|
Physiological polymerization of TDP-43 in health and disease
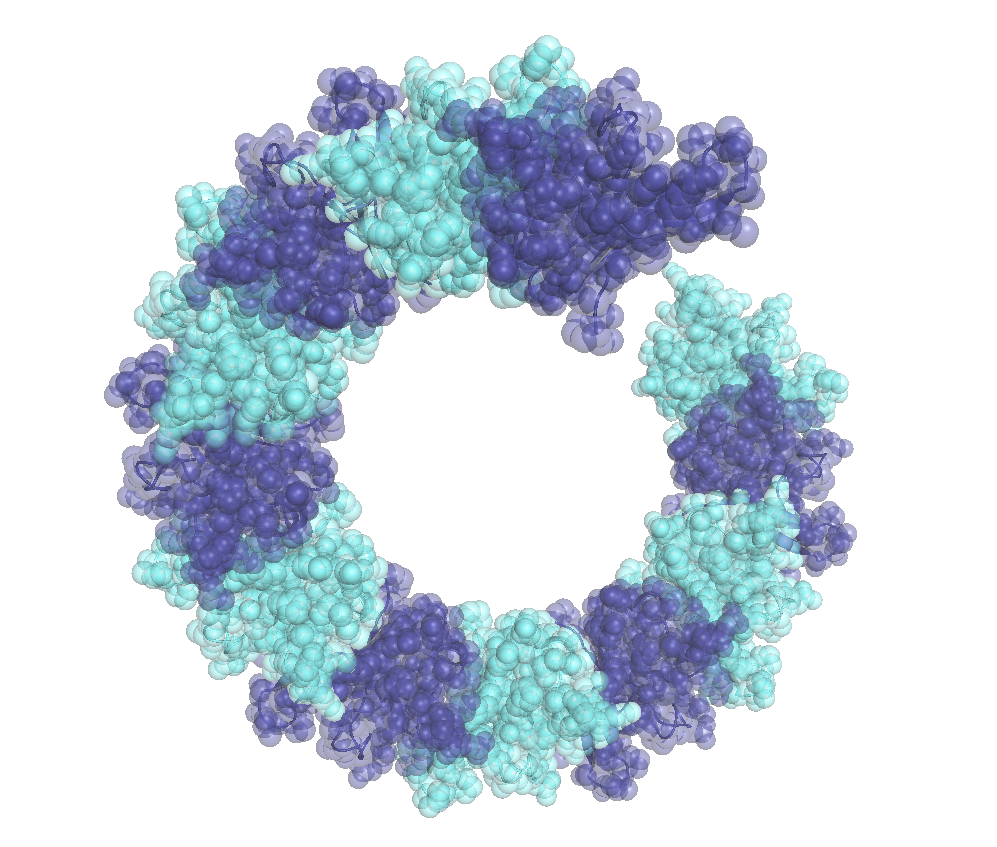
TDP-43 is a primarily nuclear RNA-binding protein, whose abnormal phosphorylation and cytoplasmic aggregation characterizes affected neurons in patients with ALS and FTD. We discovered that physiological nuclear TDP-43 in mouse and human brain forms dynamic oligomers via self-association of its N-terminal domain (NTD), which represent the functional form of the protein in vivo, since their destabilization results in loss of alternative splicing regulation of known neuronal RNA targets. Surprisingly, we found that physiological TDP-43 oligomerization antagonizes the formation of pathologic aggregates (Afroz et al, Nature Comm, 2017), potentially by the spatial separation of the low complexity regions of neighboring TDP-43 molecules (Foglieni et al, Scientific Reports, 2017). We are currently exploring the role of loss of TDP-43 physiologic oligomerization in disease. |
|
Role of cellular stress in the initiation of FUS pathology
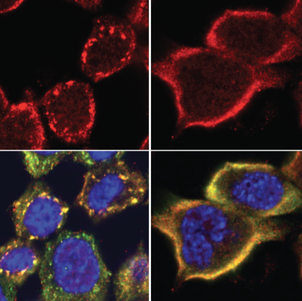
The primarily nuclear RNA-binding protein FUS forms pathological cytoplasmic inclusions in a subset of early onset ALS and FTD patients. In response to cellular stress, FUS is recruited to cytoplasmic stress granules, which are hypothesized to act as precursors of pathological inclusions. We monitored the stress-induced nucleocytoplasmic shuttling of endogenous FUS ex vivo in the mouse CNS and in human neural networks. We found that hyperosmolar, but not oxidative stress induced robust cytoplasmic translocation of neuronal FUS, with transient nuclear clearance and loss of function. Surprisingly, this reaction is independent of stress granule formation and does not occur in astrocytes, which remain unaffected in ALS/FTD-FUS (Hock et al, Cell Reports, 2018). We are currently working on identifying the molecular “switch” that controls this differential response of astrocytes versus neurons aiming to determine a possible resistance mechanism to FUS pathology. |
|
|
Molecular basis of TDP-43 disease heterogeneity
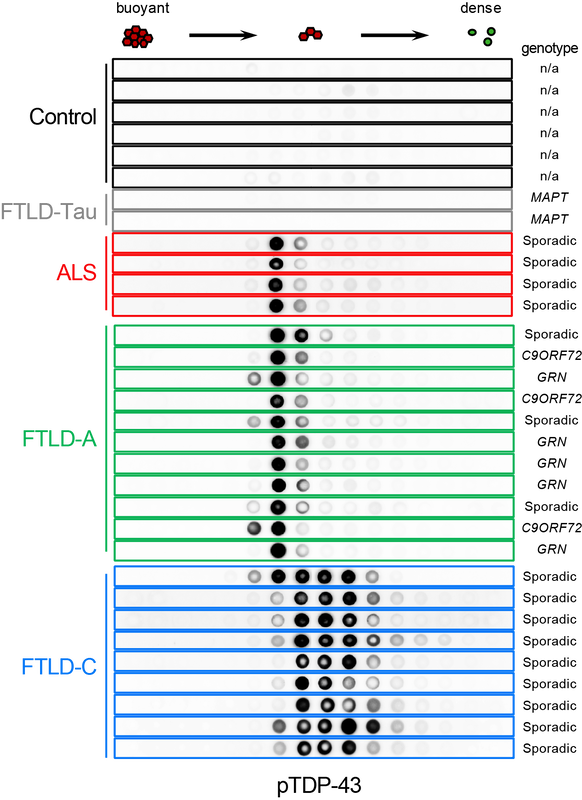
Accumulation of abnormally phosphorylated TDP-43 (pTDP-43) is the main pathology in affected neurons of people with amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). Morphological diversity and neuroanatomical distribution of pTDP-43 accumulations allowed classification of FTLD cases into at least four subtypes, which are correlated with clinical presentations and genetic causes. To understand the molecular basis of this heterogeneity, we developed SarkoSpin, a new method for biochemical isolation of pathological TDP-43. We showed that pTDP-43 extracted from brain forms stable assemblies of distinct densities and morphologies that are associated with disease subtypes. Importantly, biochemically extracted pTDP-43 assemblies showed differential neurotoxicity and seeding that were correlated with disease duration of FTLD subjects. Our data are consistent with the notion that disease heterogeneity could originate from alternate pathological TDP-43 conformations, which are reminiscent of prion strains (Laferriere et al, Nature Neurosc, 2019). Following these exciting data, we are now working towards the identification of the molecular pathways that underlie the differential propagation and toxicity of these diverse pathological TDP-43 assemblies. |
|
Functional impact of dipeptide repeat proteins in human neural networks
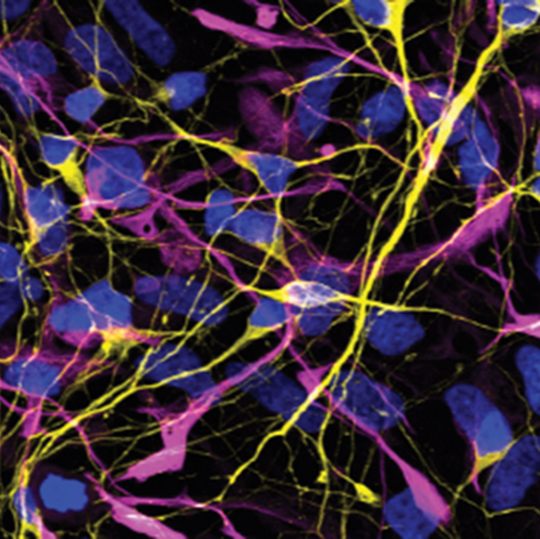
G4C2 repeat expansions in the C9ORF72 gene represent the most common cause of familial amyotrophic lateral sclerosis and frontotemporal dementia. A non-canonical type of translation of the expanded RNA produces five different dipeptide repeat proteins (DPRs), which disrupt multiple cellular processes, yet their role in disease remains controversial. While poly-GA is the most common DPR detected in patients and shows clinico-pathological association, it appears only moderately toxic in experimental models. To explore the toxicity of poly-GA in the context of mature human neurons, we generated stable, unmodified, human neural stem cells along with a robust differentiation method that yields functional, interconnected and long-lived neurons. Inducible expression of poly-GA and other DPRs in this system is used to understand their behavior and functional consequences in human neurons, as well as evaluate the efficacy of antibodies and other compounds targeting DPRs against C9ORF72 disease. |
|
|
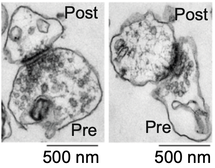
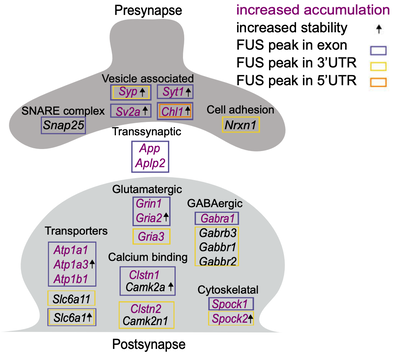
Misregulation of synaptic FUS RNA targets in disease
Mutations disrupting the nuclear localization of the RNA-binding protein FUS characterize a subset of amyotrophic lateral sclerosis patients (ALS-FUS). FUS regulates nuclear RNAs, but its role at the synapse is poorly understood. Using super-resolution imaging we determined that the localization of FUS within synapses occurs predominantly near the vesicle reserve pool of presynaptic sites. Using CLIP-seq on synaptoneurosomes, we identified synaptic FUS RNA targets, encoding proteins associated with synapse organization and plasticity. Significant increase of synaptic FUS during early disease in a mouse model of ALS was accompanied by alterations in density and size of GABAergic synapses. mRNAs abnormally accumulated at the synapses of 6-month-old ALS-FUS mice were enriched for FUS targets and correlated with those depicting increased short-term mRNA stability via binding primarily on multiple exonic sites. Our study indicates that synaptic FUS accumulation in early disease leads to synaptic impairment, potentially representing an initial trigger of neurodegeneration (Sahadevan et al, Nature Comm, 2021). We are now working on determining the molecular mechanism of synaptic RNA regulation via direct binding of FUS at the synapse. |